Mayank Murali
Computational Associate II • Broad Institute of MIT and Harvard
Computational Associate II • Broad Institute of MIT and Harvard
I'm a computational researcher specializing in statistical and computational genomics, currently working as a Computational Associate II at the Broad Institute of MIT and Harvard in the Claussnitzer Lab. My work centers on the genetics of cardiometabolic disease — building pipelines for genotyping, polygenic risk scoring, and LDSC heritability estimation, and using single-cell ATAC-seq to connect GWAS variants to transcription-factor activity in adipose tissue.
Before the Broad, I worked in the Sheynkman Lab at the University of Virginia on long-read proteogenomics, where I built Biosurfer, a tool for tracking how alternative splicing reshapes protein isoforms. I hold an M.S. in Computer Science from Penn State, where my thesis benchmarked long-read metagenome assembly tools.
View My Research
Computational Associate II · Claussnitzer Lab
Nov 2023 - Present
Building statistical genetics pipelines that connect genetic variation to cardiometabolic disease risk, from genotyping through single-cell functional genomics.
Senior Data Analyst · Sheynkman Lab
Jul 2022 - Nov 2023
Developed Biosurfer and supporting long-read proteogenomics infrastructure to study protein isoform diversity.
Graduate Teaching Assistant
Jan 2020 - Dec 2021
Led instruction for an undergraduate systems programming course.
medRxiv (preprint) · submitted to Nature Metabolism
bioRxiv (preprint), 2025.11.18.689066
American Society of Human Genetics (ASHG) Annual Meeting, Boston, MA · Poster
American Journal of Human Genetics, 111(9), 1914–1931
Broad Institute Retreat, Cambridge, MA · Poster
Current Science, 126(8), 878–881
RNA Biology, 19(1), 1228–1243
International Journal of Recent Technology & Engineering (Scopus-indexed), 8(1), 220–223
Proc. 3rd International Conference on Computing and Communications Technologies (ICCCT), IEEE
Manuscripts in preparation (co-author): Eliasen AU, Thorsen J, Dashti H, et al., Integration of genetic association and tissue-specific expression data reveals brain and non-brain genetic mechanisms of obesity (with M. Claussnitzer, J. Hirschhorn) · Díez-Obrero V, Svenstrup V, Smit RAJ, et al., Multi-omic gene prioritization within body-mass-index loci provides molecular insight into their variant-to-function translation.
M.S. thesis project: built an automated pipeline to benchmark de novo metagenome assemblers on long-read PacBio HiFi data by misassembly rate, assembly quality, and genome completeness, with contig visualization via Bandage on an HPC cluster. Advised by Prof. Mingfu Shao.
View on GitHubBuilt a Galaxy pipeline for ChIP-exo and RNA-seq analysis to identify Forkhead Box C1 (FOXC1) binding sites in mouse stem-cell data and investigate regulation of the gene.
Benchmarked unordered map, suffix array, hash table, and Bloom filter implementations against the KMC 2 algorithm for k-mer counting on the human GRCh38 and Staphylococcus aureus genomes, comparing memory usage and runtime.
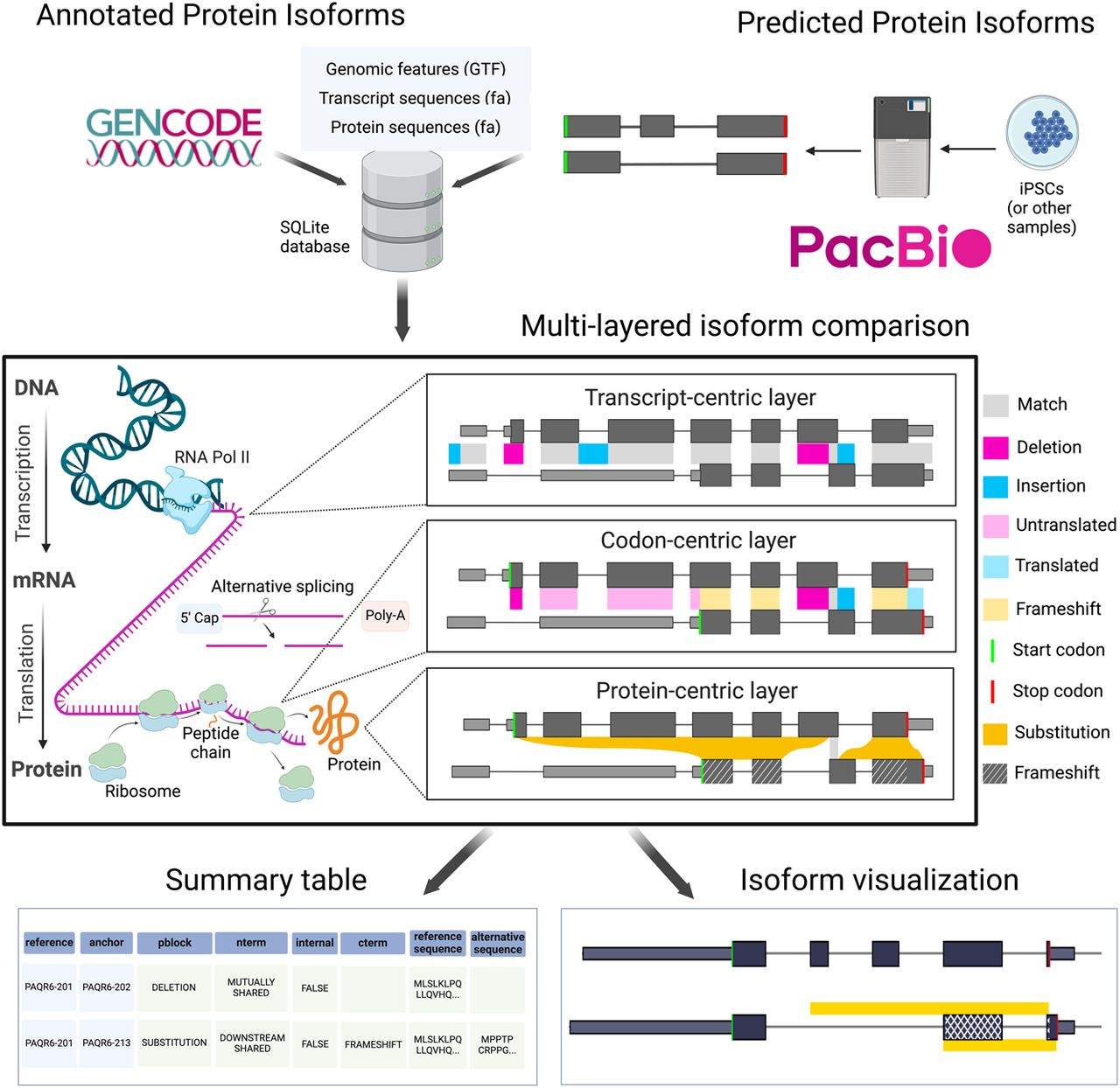
Biosurfer is a computational tool for comparing protein isoforms by tracking transcriptional, splicing, and translational variations that drive sequence differences. Analysis of 35K+ GENCODE protein pairs revealed that 70% of variable N-termini arise from alternative transcription start sites, while 72% of C-terminal changes involve frameshift-inducing splicing events. Available as a Python package.
Built genotyping, polygenic risk score (PRS), and LDSC heritability pipelines on the Terra platform for the Claussnitzer Lab's in-house biobank, and developed a framework linking single-cell ATAC-seq data to transcription-factor motif disruption in adipose tissue — connecting GWAS variants to cardiometabolic disease mechanisms. Extended these methods to large-scale population-genetics analyses of the FinnGen biobank to study the shared genetic and epigenetic basis of type 2 diabetes.
B.Tech in Computer Science & Engineering
2015 - 2019
Studied foundational computer science and engineering coursework — data structures, algorithms, DBMS, operating systems, computer architecture — alongside undergraduate research in applied IoT/ML systems.
M.S. in Computer Science · GPA 3.25/4.0
2019 - 2022
M.S. Thesis: benchmarked de novo metagenome assembly tools on long-read PacBio HiFi data, advised by Prof. Mingfu Shao.